VeloxChem: Electron Repulsion Integrals and Fock Matrix Formation on GPUs
Xin Li, PDC
Quantum chemistry offers invaluable insight into and understanding of the complexities of electronic structure and can, therefore, serve as a very powerful tool in scientific research and theoretical design of molecules and materials. The major computational complexity in quantum chemistry lies in the evaluation of the electron repulsion integrals (ERI) and the formation of the so-called Fock matrix, which formally scales as O(N4), with N being the system size. In practice, this time-consuming task can be done much more efficiently by taking advantage of the screening of the ERIs, and utilisation of GPUs can provide significant speedup.
The VeloxChem program [1] is an open-source quantum chemistry software application developed at the KTH Royal Institute of Technology (KTH), including at PDC. Recently, the VeloxChem team at KTH and PDC implemented ERI evaluation and Fock matrix formation on GPUs to further push the limit of the sizes of the systems that can be routinely studied. In our implementation, the ERIs are evaluated by the Obara-Saika recurrence relation [2], and, on top of that, the formation of the Fock matrix is done by contracting the ERIs with the density matrix. The Fock matrix can be split into two contributions due to different contracting patterns, which are called the Coulomb and the exchange contributions, respectively. To take advantage of the O(N2) scaling of the Coulomb contribution and the O(N) scaling of the exchange contribution, we implemented the Fock matrix formation based on the direct self-consistent field implementation [3] and the pre-selective screening approach [4].
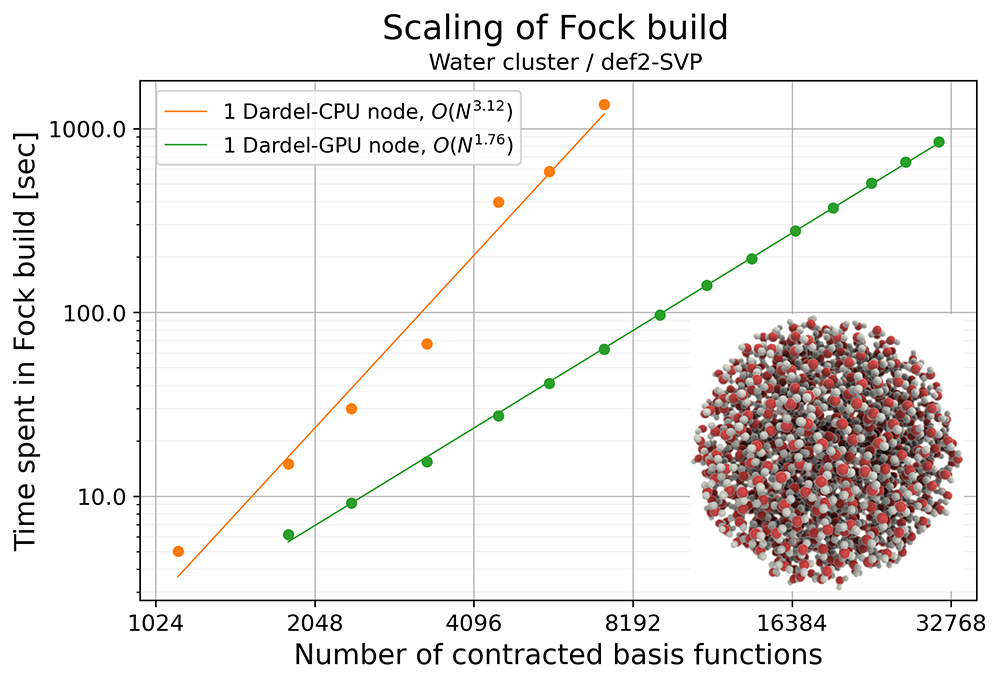
We tested the performance of the Fock matrix formation and ERI evaluation in VeloxChem by running Hartree-Fock calculations for a series of water clusters on Dardel. The largest water cluster used in the benchmark contains 3,879 atoms, and the number of contracted basis functions exceeds 31,000 with the def2-SVP basis set [5]. By plotting the time spent in Fock matrix formation with respect to the size of the system (indicated by the number of contracted basis functions), we can see that the scaling of the computational cost on GPUs is O(N1.76), which falls between the expected scaling of the Coulomb and exchange contributions. Due to such beneficial scaling, the formation of the Fock matrix for the largest water cluster took around 850 seconds on a Dardel GPU node. This opens up the possibility of routine study of large and complex chemical systems.
References
- Z. Rinkevicius, X. Li, O. Vahtras, K. Ahmadzadeh, M. Brand, M. Ringholm, N. H. List, M. Scheurer, M. Scott, A. Dreuw, P. Norman, “VeloxChem: a Python-driven density-functional theory program for spectroscopy simulations in high-performance computing environments”. WIREs Comput. Mol. Sci. 2020, 10:e1457. doi.org/10.1002/wcms.1457
- S. Obara, A. Saika, “Efficient recursive computation of molecular integrals over Cartesian Gaussian functions”. J. Chem. Phys. 1986, 84, 3963-3974, doi.org/10.1063/1.450106
- I.S. Ufimtsev, T. J. Martinez, “Quantum Chemistry on Graphical Processing Units. 2. Direct Self-Consistent-Field Implementation”. J. Chem. Theory Comput. 2009, 5, 1004-1015, doi.org/10.1021/ct800526s
- J. Kussmann, C. Ochsenfeld, “Pre-selective screening for matrix elements in linear-scaling exact exchange calculations”. J. Chem. Phys. 2013, 138, 134114, doi.org/10.1063/1.4796441
- F. Weigend, R. Ahlrichs, “Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy”. Phys. Chem. Chem. Phys., 2005, 7, 3297-3305, doi.org/10.1039/b508541a