Co-design Efforts for Major Performance Improvements in GROMACS GPU-Accelerated Parallelisation
Szilárd Páll, PDC, & Alan Gray, NVIDIA
The GROMACS open-source molecular dynamics (MD) code is among the early high-performance computing (HPC) scientific codes to make efficient use of graphics processing unit (GPU) accelerators with its GPU support having been available for more than decade. The heterogeneous parallelisation of the GROMACS code has gone through a gradual evolution, from offloading only the most compute-intensive parts of a simulation to increasingly moving most computation from the CPUs to the GPUs to make efficient use of modern accelerator-based systems.
Collaboration between academic and industry partners, including hardware vendors, has been an important driver in terms of hardware support for GROMACS. The GROMACS team’s close collaboration with NVIDIA on GPU parallelisation has been of great benefit from the very early days of GPU support in relation to GROMACS.
Funding through a Swedish Foundation for Strategic Research (SSF) Swedish Exascale Computing Initiative (SECI) project (from 2016 to 2022) enabled PDC-based researchers and members of the GROMACS team to focus on improving molecular dynamics algorithms for exascale HPC architectures. Thanks to this funding and shared objectives, we established a co-design collaboration between the GROMACS team and NVIDIA with a long-term goal to advance molecular dynamics algorithms and redesign the GROMACS parallelisation, adapting it to modern GPU-accelerated architectures. At the same time, this co-design effort was also successful at providing feedback to the NVIDIA software teams about the requirements of molecular dynamics algorithms and GROMACS implementation.
One of the major focuses of this co-design collaboration has been improving the strong scaling of molecular simulations on multi-GPU machines. Portable implementations have been developed in GROMACS, including GPU-resident parallelisation with direct-GPU communication [1]. Research efforts have also focused on more efficient task scheduling on GPUs and explored new ways to express fine-grained parallelism in molecular simulation by employing task graph-based scheduling [2].
The direct GPU communication layer, which allows making efficient use of high-performance interconnects and helps avoid staging data movement through the CPU, was extended with GPU-aware MPI support in the 2022 GROMACS release. Building on this, recent efforts focused on removing a strong scaling bottleneck which limited the scalability of GROMACS on recent HPC architectures where most of the computational power is provided by GPUs.
The particle mesh Ewald (PME) algorithm is used most commonly to compute long-range electrostatics interactions in MD. This relies on fast Fourier transform (FFT) operations, which are hard to parallelise and scale, especially the typically small transforms required for biomolecular MD. While computing the short-range interactions could be scaled efficiently across multiple GPUs and multiple nodes with GROMACS, until recently the entire PME computation had to use a single (although dedicated) GPU which would quickly limit the scaling of such simulations (typically to 4 to 8 GPUs). Alternatively, the multi-node simulation could use CPUs for the entire PME computations, but this also has limited performance and scaling on modern architectures.
To lift this limitation in our co-design project, we have developed a PME decomposition algorithm better suited for GPUs (which relies on grid overlap reduction, instead of particle redistribution prior to spreading to the grid) and added support for distributed FFT computations using both the portable HeFFTe [3] library, as well as the recently introduced cuFFTmp library [4]. The former allows portability, while the latter is a more performant vendor library, which has been optimised with the GROMACS and MD use-case as one of its targets.
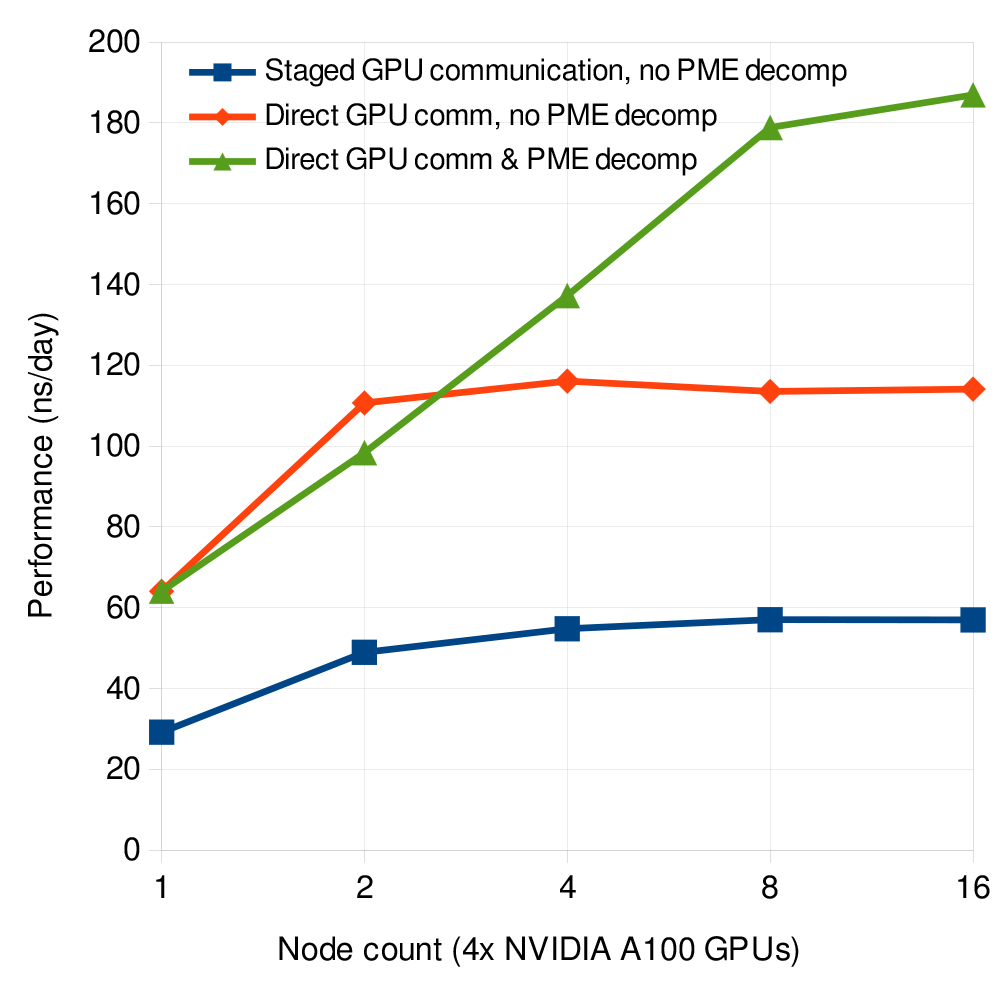
Thanks to the PME GPU decomposition, the recent 2023 release of GROMACS is able to distribute PME computation across multiple GPUs within or across compute nodes, thereby lifting a major scaling limitation and offering major performance improvements in simulations typically from 6 to 8 GPUs.
References
- S. Páll, A. Zhmurov, P. Bauer, M. Abraham, M. Lundborg, A. Gray, B. Hess & E. Lindahl, Heterogeneous Parallelization and Acceleration of Molecular Dynamics Simulations in GROMACS. The Journal of Chemical Physics 153, no. 13 (7 October 2020): 134110. doi.org/10.1063/5.0018516
- developer.nvidia.com/blog/a-guide-to-cuda-graphs-in-gromacs-2023
- heFFTe icl.utk.edu/fft
- cuFFTmp docs.nvidia.com/hpc-sdk/cufftmp/index.html
- NVIDIA Selene www.top500.org/system/179842