GROMACS 2023: Readiness on the AMD GPU Heterogeneous Platform
Szilárd Páll, PDC, and Andrey Alekseenko, Scilifelab & Department of Applied Physics, KTH
GROMACS, the widely used open-source molecular dynamics package, is one of the major high-performance computing (HPC) research codes which has long supported heterogeneous graphics processing unit (GPU) acceleration. GROMACS users have benefitted from utilising GPUs for nearly a decade.
Molecular dynamics research using GROMACS is one of the significant workloads on the Dardel system at PDC, as well as on the LUMI system in Finland. CPU performance – both absolute and strong scaling on CPU-only AMD platforms (including Dardel’s CPU partition and LUMI-C) – has been excellent. At the same time, the GROMACS codebase has been being prepared for the increasingly heterogeneous and diverse HPC platforms through algorithmic, parallelisation, and portability efforts, which were discussed in the 2022 no.1 edition of this newsletter (see www.pdc.kth.se/about/publications/pdc-newsletter-2022-no-1/preparing-gromacs-for-heterogeneous-exascale-hpc-systems-1.1174906 ). Last year, the heterogeneous parallelisation and SYCL portability layer targeting AMD GPUs were enhanced in a number of key areas in preparation for providing broad support for these new accelerator-based HPC resources in the official 2023 GROMACS release [1,2], which was released in February 2023.
The GROMACS team has implemented performance optimisations targeting the AMD CDNA2 architecture at the heart of the MI250X accelerators (which are used, for example, in the GPU partition of Dardel) to improve the performance of key compute kernels. This made significant performance gains possible (with improvements ranging from 1.2 to 2.0 times previous performance), but some peculiarities of the platform proved to be challenging to address in a portable way. For example, the peak single-precision floating point throughput requires the use of packed math operations on the AMD CDNA2 architecture [3], but the AMD compiler was not able to generate such instructions, while the manual code transformations that were required were difficult to integrate into the multi-platform SYCL kernels without leading to excessive complexity. Hence, such complex optimisations have been postponed. We have reported these issues to the vendor and hope such manual architecture-specific optimisations will not be necessary with future compilers.
The GROMACS code relies on its SYCL portability backend for AMD support using the hipSYCL application programming interface (API) [4], which adds an abstraction layer on top of the base AMD ROCm stack. Due to the high iteration rates (typically less than 1 millisecond) and many small tasks characteristic to molecular dynamics and GROMACS, this workload puts a lot of pressure on the GPU runtime and overheads accumulate [5]. Therefore, the team has focused on identifying and eliminating such overheads. On the one hand, we have reported ROCm/HIP issues and got fixes for them. On the other hand, we have identified limitations of the SYCL abstraction layer and worked closely with the hipSYCL team to reduce related overheads. This motivated certain custom programming model extensions to reduce scheduling overheads which have made a significant impact (up to 25% reduction in wall-time) on the sub-millisecond iteration rate GROMACS simulations. At the same time, these improvements have also become important use-cases for the direction the SYCL standard should progress towards. The team are contributing to this through our SYCL Advisory Panel membership and its feedback process.
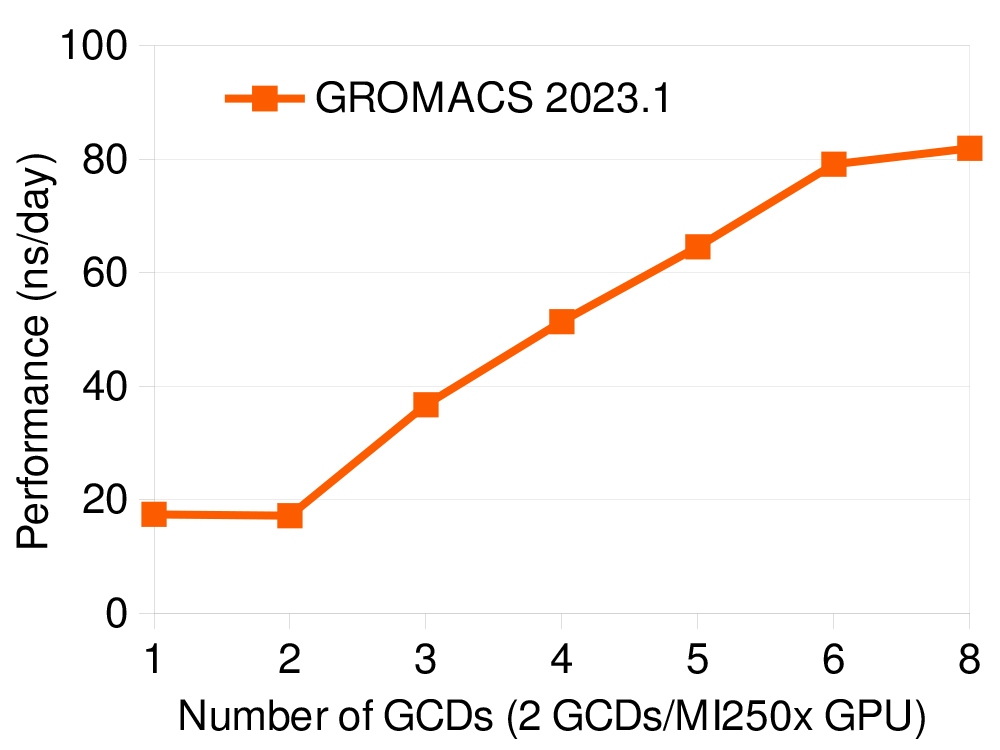
The GROMACS team has also worked on improving feature-completeness which, in the latest release, is on par with the most mature CUDA GPU backend when it comes to single-GPU, and multi-GPU/multi-node simulations, including using GPU-aware MPI. Therefore, key features of the GROMACS parallelisation, the GPU-resident mode and direct GPU communication, are supported in the latest release, allowing the best use of the accelerator-dense AMD GPU platform and its intra- and inter-node interconnects. The production code has gone through stabilisation and the official GROMACS 2023 release shows good performance, as shown in the graph above and in the table below, for a few representative benchmark systems, and is typically within 20-25% of the performance of AMD’s own GROMACS fork.
Benchmark system |
Performance (ns/day) |
|---|---|
| rnase_cubic (24k atoms) | 630.0 |
| adh_cubic (134k atoms) | 215.0 |
| STMV (1M atoms) | 17.9 |
Despite the small size of the team working on these features, major leaps have been made possible by early investments into portable algorithms (which was discussed in the PDC newsletter mentioned earlier; for additional details, see [6]) and portable standards-based APIs. In addition, close collaboration between GROMACS developers at PDC and other departments at the KTH Royal Institute of Technology was an important contributing factor, as was the contribution of portable algorithms and code by a wide range of hardware vendors, including Intel (through the KTH-Intel OneAPI Centre of Excellence) and NVIDIA (as part of the GROMACS-NVIDIA co-design collaboration).
References
- GROMACS 2023 release highlights manual.gromacs.org/2023/release-notes/2023/major/highlights.html
- bioexcel.eu/webinar-whats-new-in-gromacs-2023-2023-04-25
- AMD CDNA2 white paper. www.amd.com/system/files/documents/amd-cdna2-white-paper.pdf
- hipSYCL project page. github.com/illuhad/hipSYCL
- A. Alekseenko and S. Páll. 2023. Comparing the Performance of SYCL Runtimes for Molecular Dynamics Applications. In Proceedings of the 2023 International Workshop on OpenCL (IWOCL ’23). Association for Computing Machinery, New York, NY, USA, Article 6, 1–2. doi.org/10.1145/3585341.3585350
- S. Páll, A. Zhmurov, P. Bauer, M. Abraham, M. Lundborg, A. Gray, B. Hess & E. Lindahl. “Heterogeneous Parallelization and Acceleration of Molecular Dynamics Simulations in GROMACS.” The Journal of Chemical Physics 153, no. 13 (7 October 2020): 134110. doi.org/10.1063/5.0018516